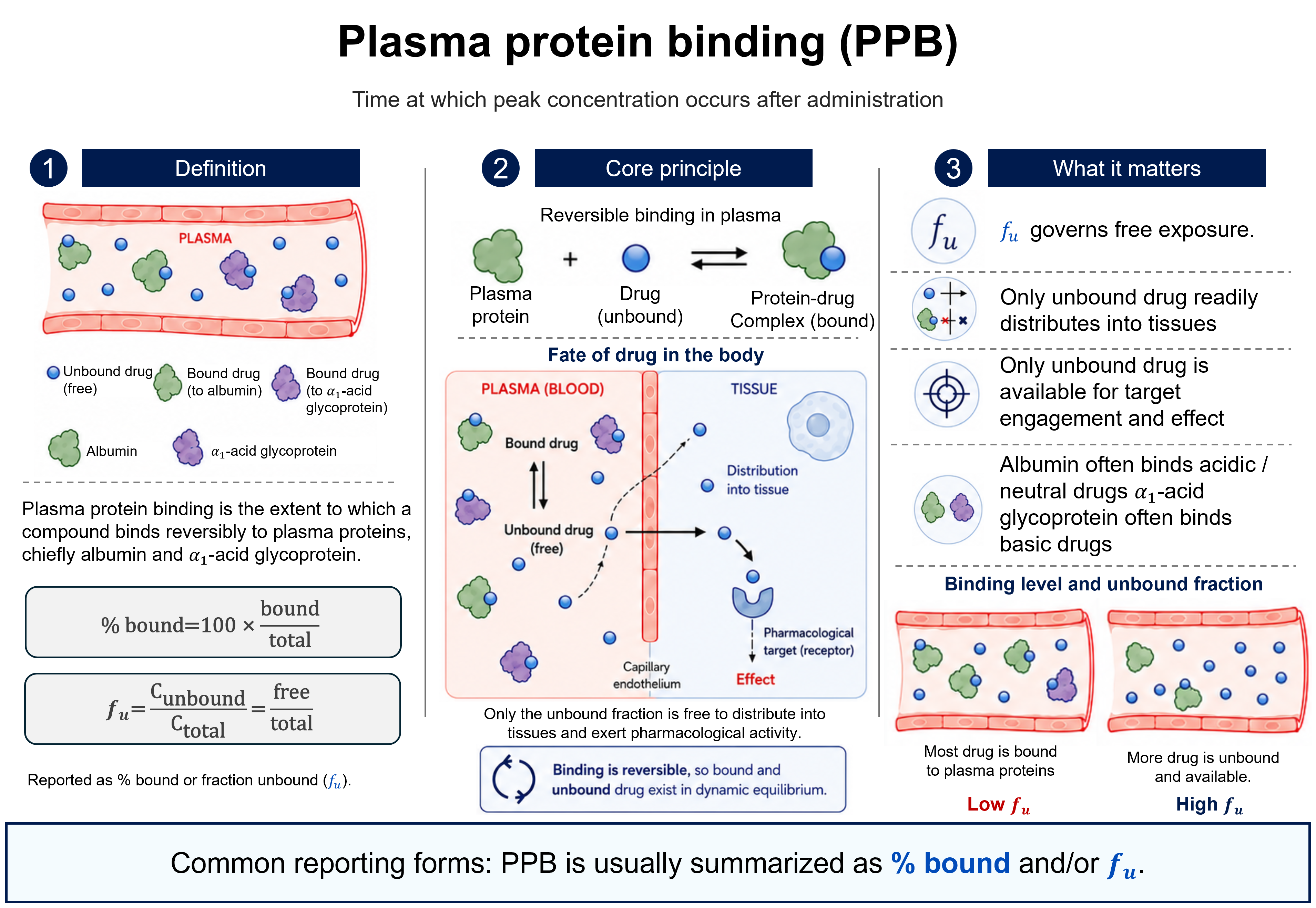
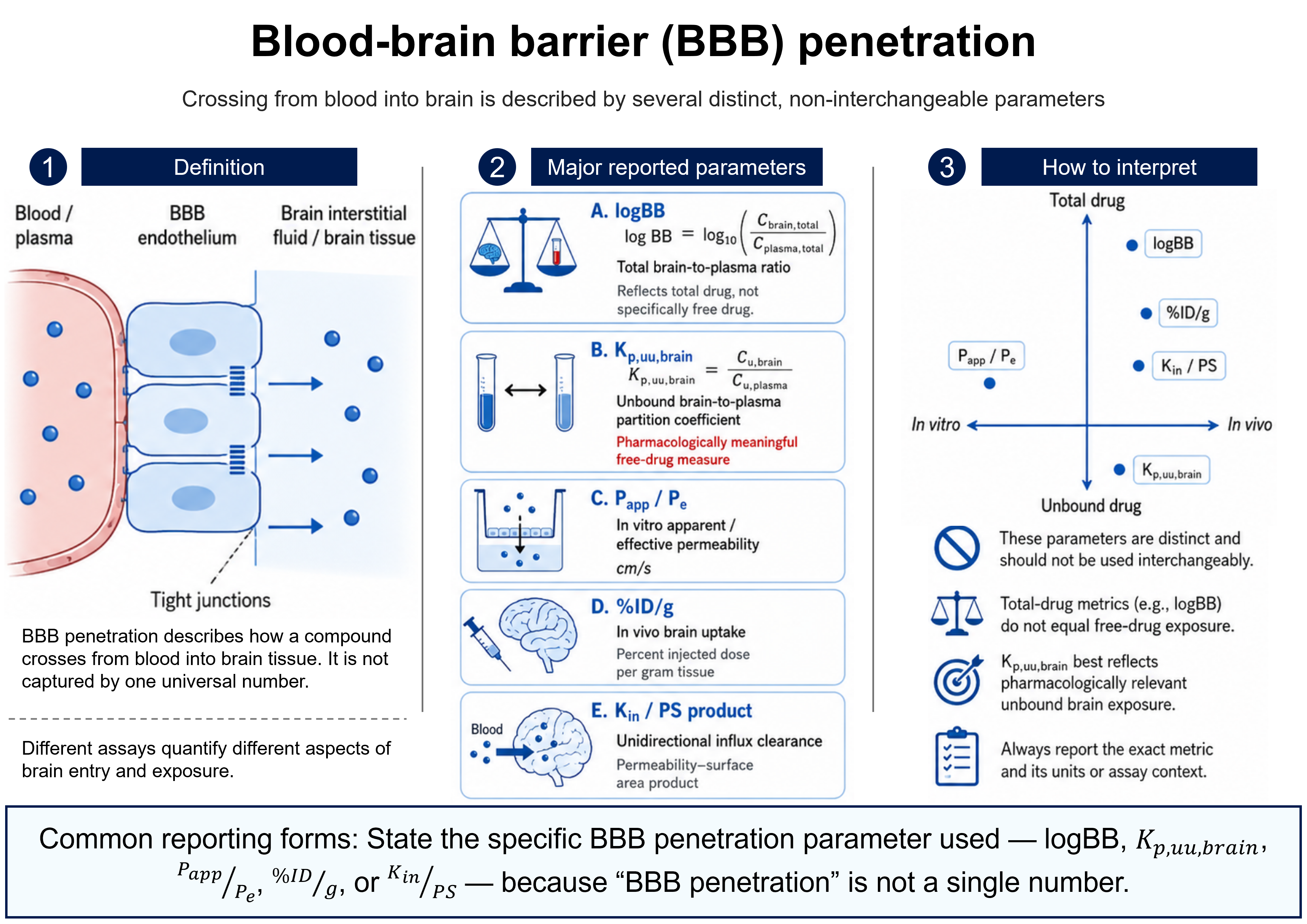
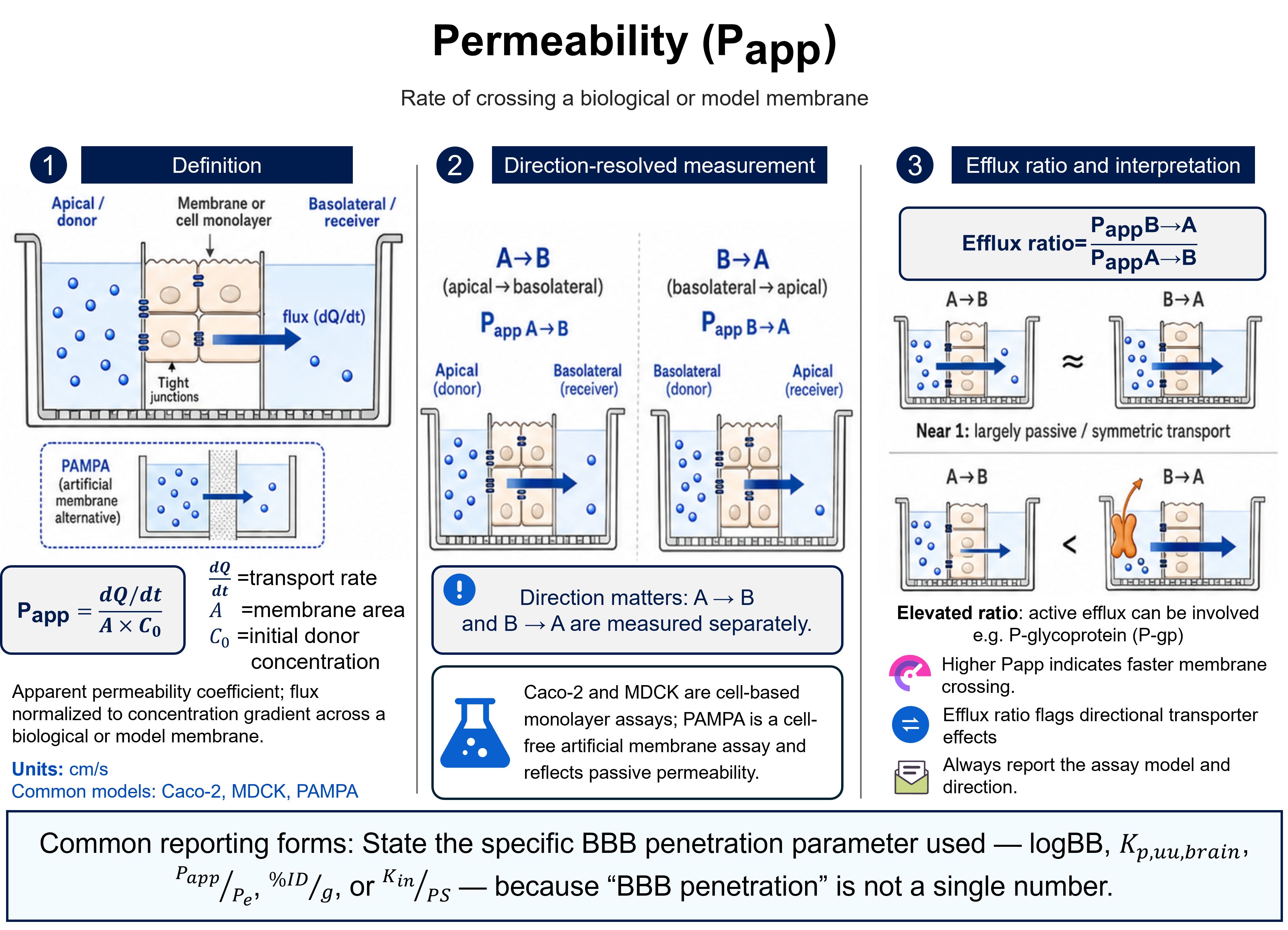
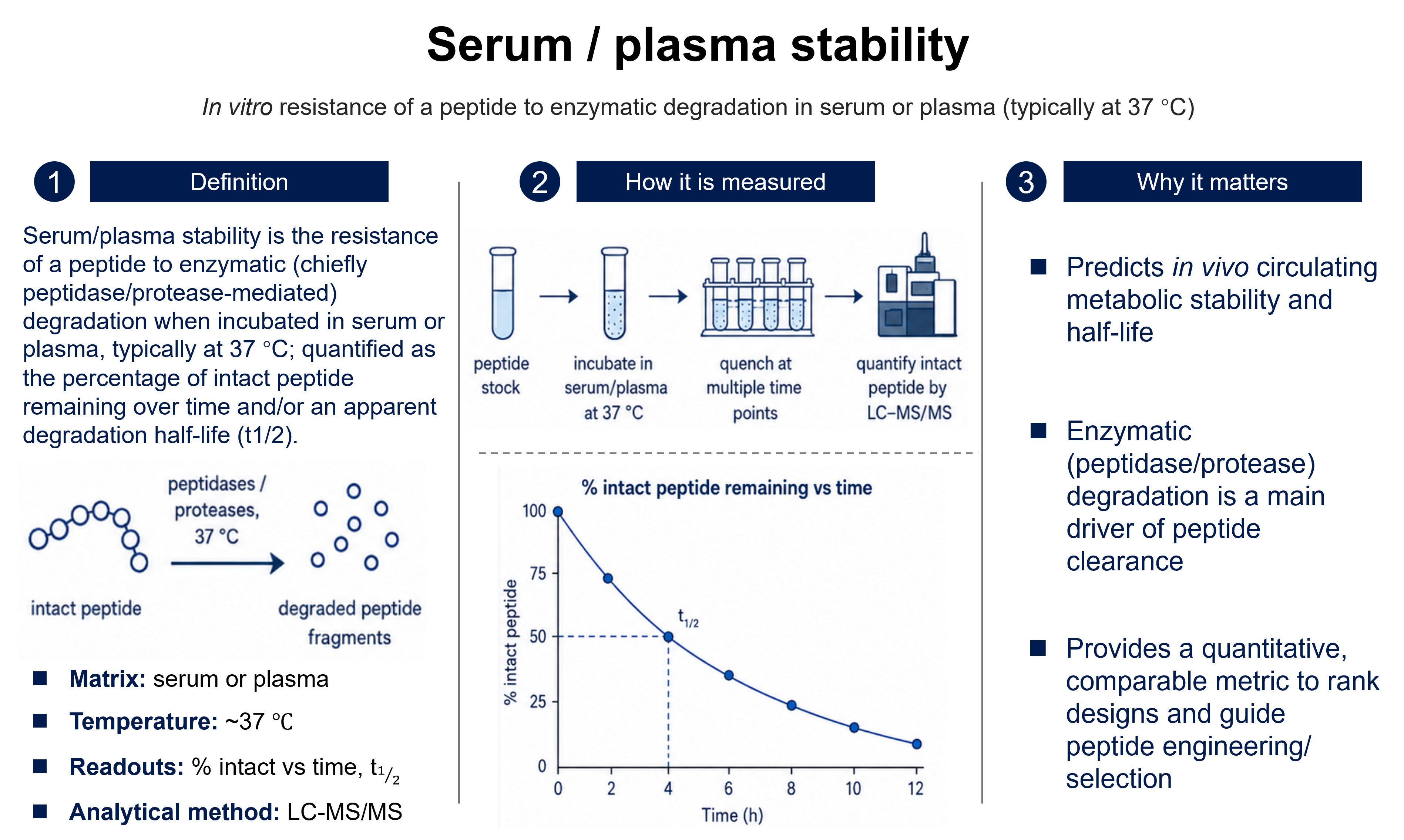
Bioavailability
Ffraction or %Bioavailability is the rate and extent to which the active moiety is absorbed from a dosage form and becomes available in the systemic circulation. Absolute bioavailability (F) is the dose-corrected fraction of drug reaching the circulation relative to intravenous administration (IV defined as 100%); relative bioavailability compares two non-intravenous routes or formulations. It is expressed as a fraction or percent.
How to read it
- Always distinguish absolute (vs IV) from relative (vs another non-IV reference) bioavailability.
- Values are species- and route-specific.
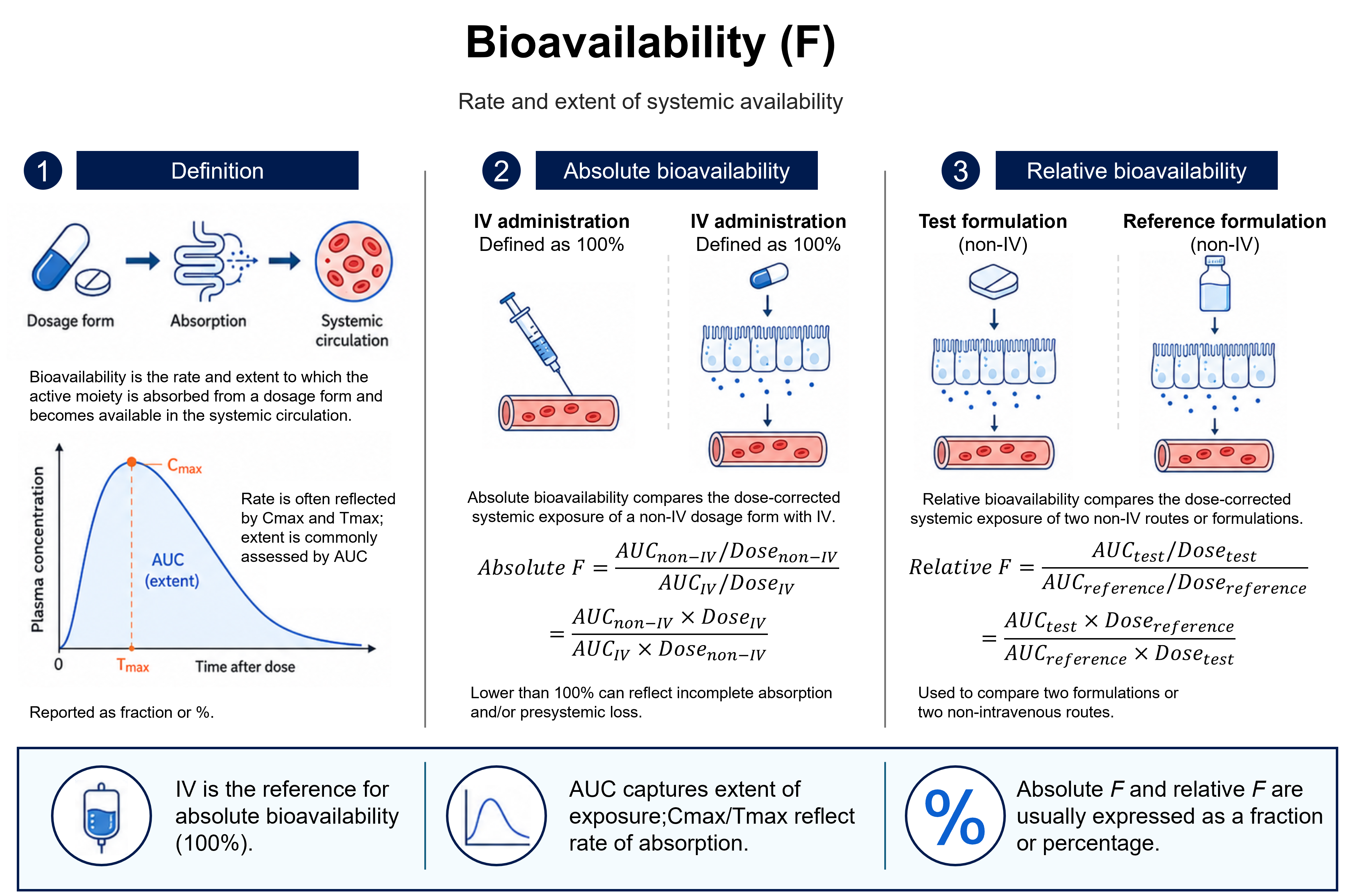
Reading the figure
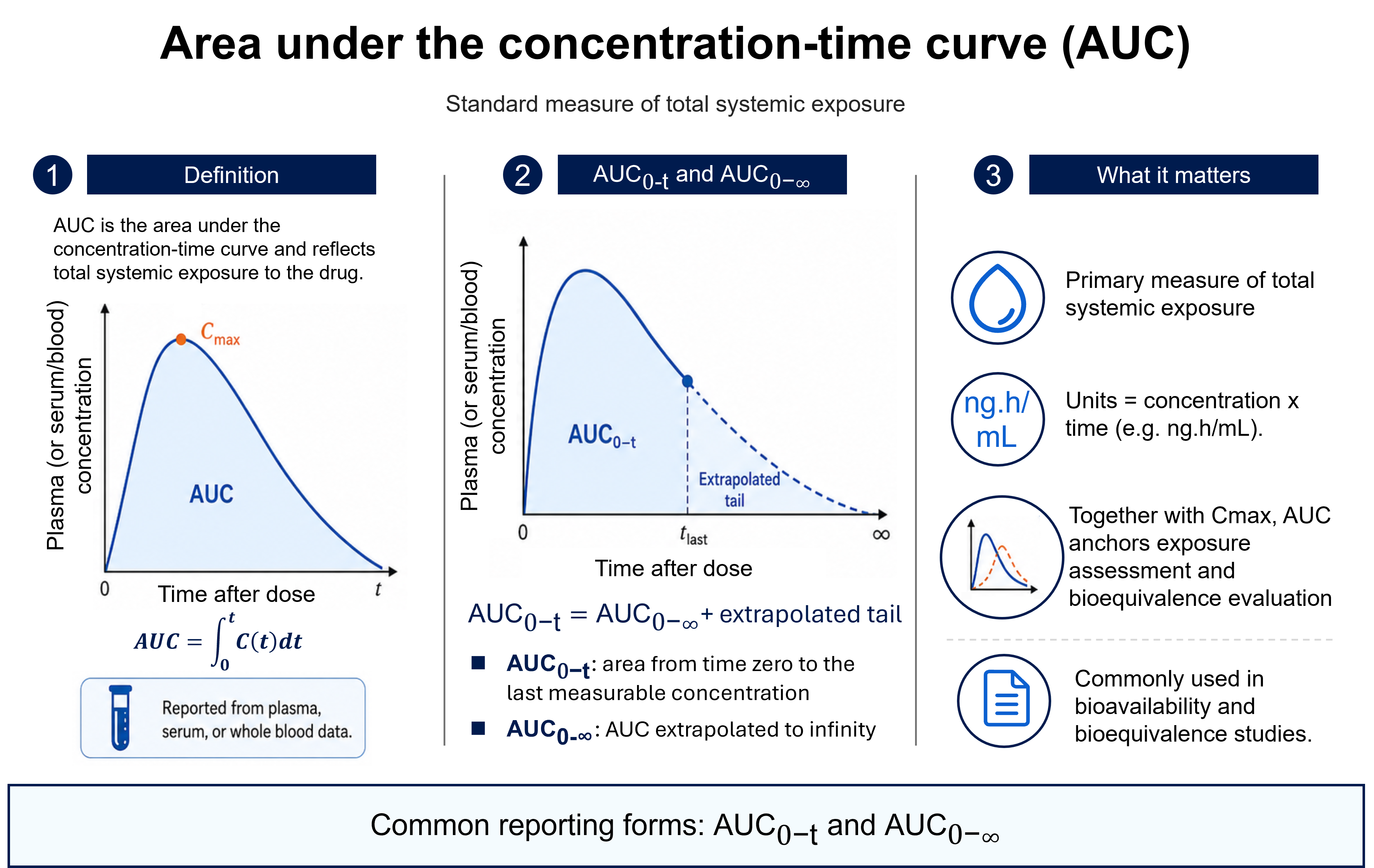
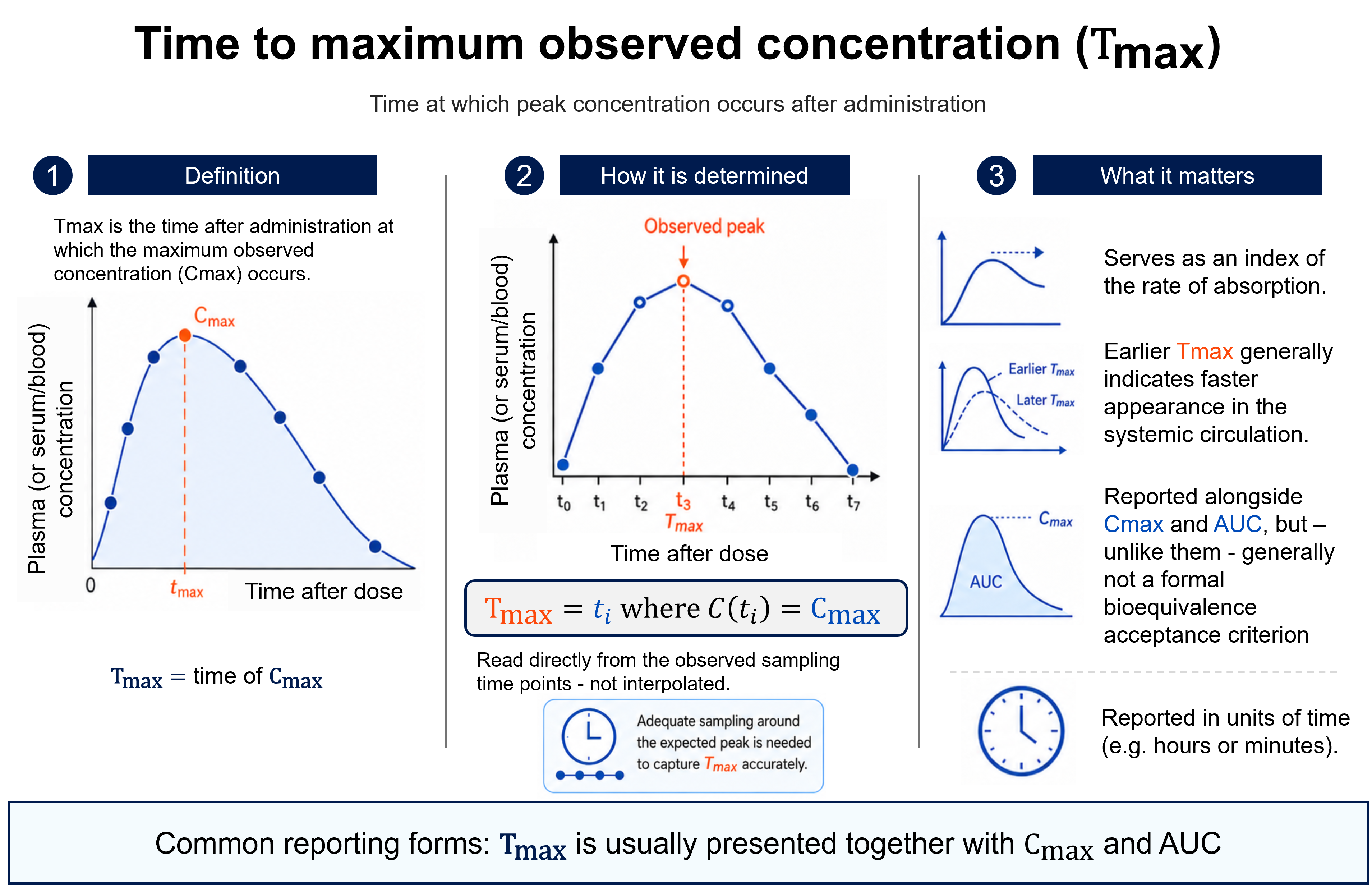
The figure follows a drug from its dosage form, through absorption, into the systemic circulation, and shows how that exposure is quantified. On the plasma concentration–time curve, the peak concentration (Cmax) and the time at which it occurs (Tmax) index the rate of absorption, while the area under the curve (AUC) measures the extent of exposure — the total amount of active moiety that actually reaches the circulation.
Panel 2 defines absolute bioavailability. Because an intravenous dose enters the bloodstream completely, it is taken as the 100% reference, and any non-intravenous route (oral, subcutaneous, …) is compared against it after correcting for dose — so F is the dose-normalised ratio of the two AUCs. A value below 100% reflects drug that never reaches the circulation intact, through incomplete absorption across the gut wall and/or presystemic (first-pass) loss in the gut and liver.
Panel 3 contrasts this with relative bioavailability, which compares two non-intravenous formulations or routes with each other — for example a new tablet against a reference oral solution — rather than against IV. The same dose-corrected AUC ratio is used, so the result expresses how the test formulation performs against the chosen reference, not against the theoretical maximum.
References
- [1]FDA. Bioavailability Studies Submitted in NDAs or INDs — General Considerations: Guidance for Industry. 2022.
- [2]EMA. Guideline on the Investigation of Bioequivalence (CPMP/EWP/QWP/1401/98 Rev. 1). 2010.
- [3]Rowland M, Tozer TN. Clinical Pharmacokinetics and Pharmacodynamics: Concepts and Applications. 5th ed. Wolters Kluwer; 2019.
- [4]Price G, Patel DA. Drug Bioavailability. In: StatPearls. NCBI Bookshelf; 2023.
- [5]Drucker DJ. Advances in oral peptide therapeutics. Nat Rev Drug Discov. 2020;19(4):277–289.
![Maximum observed concentration (Cmax): the highest drug concentration observed in plasma/serum/blood after administration, read at the apex of the concentration–time curve at time tmax. Cmax is the largest of the measured concentrations (Cmax = max[C(ti)]) — an observed, not interpolated, value requiring adequate sampling around the peak. It indexes peak systemic exposure, reflects both the rate and extent of absorption, is reported in concentration units (e.g. ng/mL), and together with AUC anchors bioequivalence assessment.](/explanation/cmax.png)

![Lipophilicity (logD / logP): how a compound distributes between two immiscible phases, classically n-octanol (organic) and water (aqueous); higher values mean greater hydrophobicity. logP is the IUPAC partition coefficient of a single neutral species, logP = log10([neutral]octanol/[neutral]water), and is pH-independent. logD is the distribution coefficient of all species (neutral + ionized) at a stated pH, logD(pH) = log10([all species]octanol/[all species]water); because ionized forms partition preferentially into water, logD is pH-dependent and the measurement pH must be reported (e.g. logD7.4). For non-ionizable compounds logD ≈ logP; for ionizable compounds the two differ. Both increase with hydrophobicity.](/explanation/lipophilicity.png)